






Mönckeberg’s arteriosclerosis (MA) is a pathological process involving dystrophic calcification of the tunica media layer of the arterial wall due to deposition of calcium hydroxyapatite crystals, or metastatic and metabolic calcification due to osteoblastic transformation of vascular smooth muscle cells, enhancing deposition of calcium hydroxyapatite crystals. It involves many factors including phosphorus activation of the PiT1 receptor, bone morphogenetic proteins 2 and 4, endogenous 1,25 dihydroxyvitamin D and vascular calcification activating factors. MA is possibly an expression of dysregulated osteogenic commitment of vascular progenitors, and so can lead to cardiovascular morbidity in young people who do not have known risk factors.
Active calcification with bone formation results from osteogenic differentiation of resident or circulating calcifying vascular cells. Ectopic osteogenesis involves the deposition of matrix vesicles, which are cell products that act as sites for calcium hydroxyapatite crystal precipitation. The vesicles may originate from distant sites that reach the vascular matrix as substrate circulating nucleation complexes. The content of the vesicle and the composition of the microenvironment influence whether matrix vesicles are involved with calcium deposition. Another theory points to ineffective efferocytosis, where defective phagocytic clearance of apoptotic bodies by macrophages leads to accumulation of necrotic debris promoting calcium precipitation on the debris.1
Vascular calcification is accelerated in certain chronic inflammatory conditions, such as diabetes, chronic kidney disease and Paget’s disease, and in people who are taking vitamin K antagonists such as warfarin. It is now accepted that vascular calcification is not a passive phenomenon, but it is regulated by complex, active mechanisms.2,3
It has been found that vitamin K plays a crucial role in the regulation of ectopic calcifications by reversing vascular calcification. Vitamin K is an essential co-factor for the gamma-glutamyl-carboxylase enzyme, which is responsible for activation of 17 vitamin K-dependent proteins that have the ability to bind calcium.4,5
Chronic kidney disease is a cardiovascular risk factor because it causes pathological changes in the vasculature and bone and mineral disorders; these are manifested by abnormalities of calcium, phosphorus, parathyroid hormone and vitamin D, renal osteodystrophy and extra-osseous calcification of soft tissues and blood vessels.6,7
MA is diagnosed radiographically, as areas of linear calcifications in soft tissue. The linear calcifications are referred to as ‘rail tracking’, or ‘tramlines’ when the affected vessel is viewed longitudinally. In most cases, MA shows the clinical criteria of calciphylaxis (calcification of the tunica media, small vessel mural calcification with endovascular fibrosis, extravascular calcification, and vascular thrombosis, leading to tissue ischaemia).
The term ‘Mönckeberg’s calcification’ has also been used to describe vascular calcinosis not related to chronic kidney disease in rare conditions, such as pseudoxanthoma elasticum (PXE) and the generalised arterial calcification of infancy (Keutel syndrome). In the majority of cases, MA is clinically asymptomatic, but it has been described as an independent risk factor for cardiovascular diseases and it commonly affects peripheral and coronary arteries.8 In a patient presenting with peripheral vascular disease of undefined aetiology and without any known risk factors, Mönckeberg’s calcification, although rare, could be the cause of peripheral vasculopathy.
In a study to determine population-based estimates of peripheral artery disease (PAD), critical limb ischaemia and MA, ankle–brachial pressure index (ABPI) was measured, with an ABPI <0.9 indicating PAD.9 MA prevalence was calculated for two ABPI measurements >1.3 and >1.5. Using ABPI >1.5, MA was present in 1.1% and 0.5% of men and women, respectively, but at ABPI >1.5, prevalence was 13.3% for men and 6.9% for women. However, its aetiology remains unknown.10
MA affects diverse arterial areas that have been documented in case reports (Table 1). Most of the reported cases show the clinical criteria of calciphylaxis – calcification of the tunica media, small vessel mural calcification with endovascular fibrosis, extravascular calcification and vascular thrombosis, leading to tissue ischaemia.
In the following case report, we focus on a pure medial calcification as a separate pathological entity, which represents a rare case of ischaemia that can give rise to symptoms in early life and could be limb and life-threatening.
Case Report
The patient was a 22-year-old man with an average build. He had repeated muscle cramps and fatigue with heavy exercise that was relieved by rest. After initial reports, the condition progressed and he developed bilateral claudication pain with increased intensity and duration with regular exercise, in addition to early muscle atrophy.
The patient had no history of trauma, diabetes or hypertension. He was a non-smoker and had never taken vitamin K antagonist therapy. His only reported treatment was regular use of non-steroidal anti-inflammatory drugs for pain.
On physical examination, there were no symptoms or signs of lumbar disc prolapse and an MRI scan of the lumbosacral vertebrae was normal. He had palpable bilateral femoral pulses, non-palpable popliteal and distal pulsations. His ankle–brachial index could not be assessed and a picture of progressive muscle wasting was evident, but there was no tissue necrosis or impending ischaemic gangrene.
Laboratory investigations showed a normal full blood count. Haemoglobin was 13.5 g/dl, total white cell count was 7,600/μl, with neutrophils 60%, lymphocytes 25%, monocytes 10% and normal platelet count. Serum creatinine was 0.8 mg/dl. His lipid profile showed triglycerides 4.15 mmol/l, LDL 3.2 mmol/l, total cholesterol 4.4 mmol/l and HDL 0.9 mmol/l. Rheumatoid and antinuclear factors were non-reagent. Cardiolipin antibodies immunoglobulin (Ig) M and IgG were negative. Cryoglobulins, hepatitis B and C were also negative. ESR and C-reactive protein were slightly elevated (60 mm/h and 95.7 mg/l). Protein C was 56% and protein S was 75%. Calcium in urine over 24 hours was 431.42 mg/dl, outside the normal range of 100–300 mg/dl. Parathyroid hormone was 98 pg/ml, slightly higher than the normal range of 16–87 pg/ml. ECG, echocardiogram and neurology were normal. A parathyroid scan was negative for adenoma or hyperplasia.
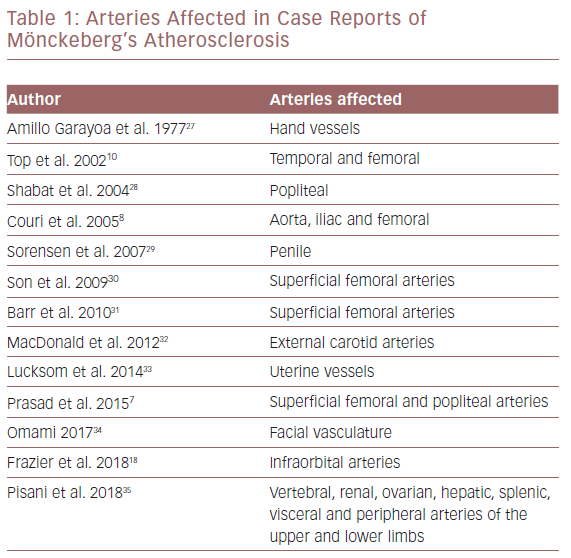
An X-ray showed extensive diffuse calcification, with some dilatation and tortuosity involving most of the arterial vascular tree of both lower limbs, indicating Mönckeberg’s medial calcific sclerosis (Figure 1).
A duplex ultrasound detected biphasic flow over both common femoral arteries and could not detect any flow from the deep femoral arteries down to the pedal arteries. This is suggestive of medial calcification.
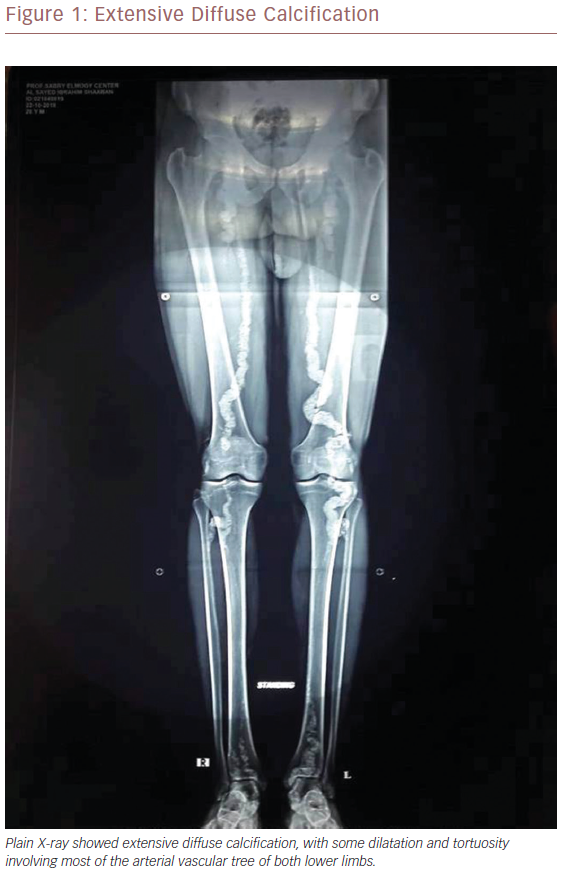
CT angiography was performed to exclude thromboembolic events or popliteal artery diseases. It revealed patent distal abdominal aorta and both common iliacs, and common femoral arteries bilaterally. Multiple large plaques of calcifications were seen, affecting the middle and distal superficial femoral artery (SFA) as well as the popliteal and deep femoral artery on both sides, with 90% stenosis of the lumen at the distal third of the SFA on both sides.
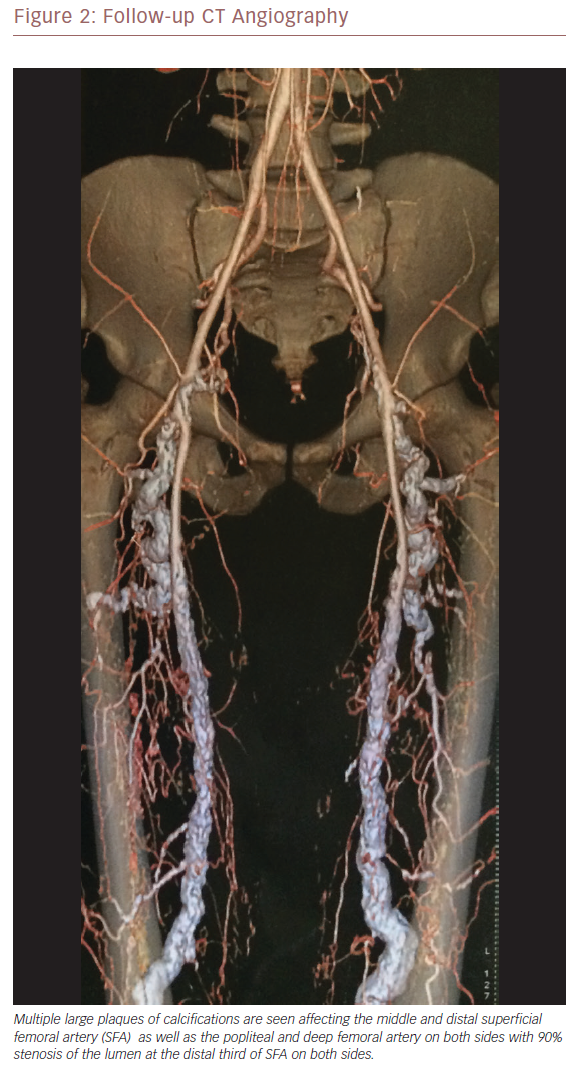
The infra-genicular region on both sides showed multiple large plaques of calcification affecting the tibioperoneal trunk and proximal anterior tibial arteries and total occlusion of the distal part of the anterior tibial arteries on both sides, with severely tortuous and dilated lower posterior tibial arteries. These findings were bilateral with a mirror image. A diagnosis of MA as a variant presentation of PXE was made at this point. He was started on cilostazol 100 mg twice daily and sevelamer hydrochloride, a non-calcium containing phosphate binder – the standard therapy for this condition.
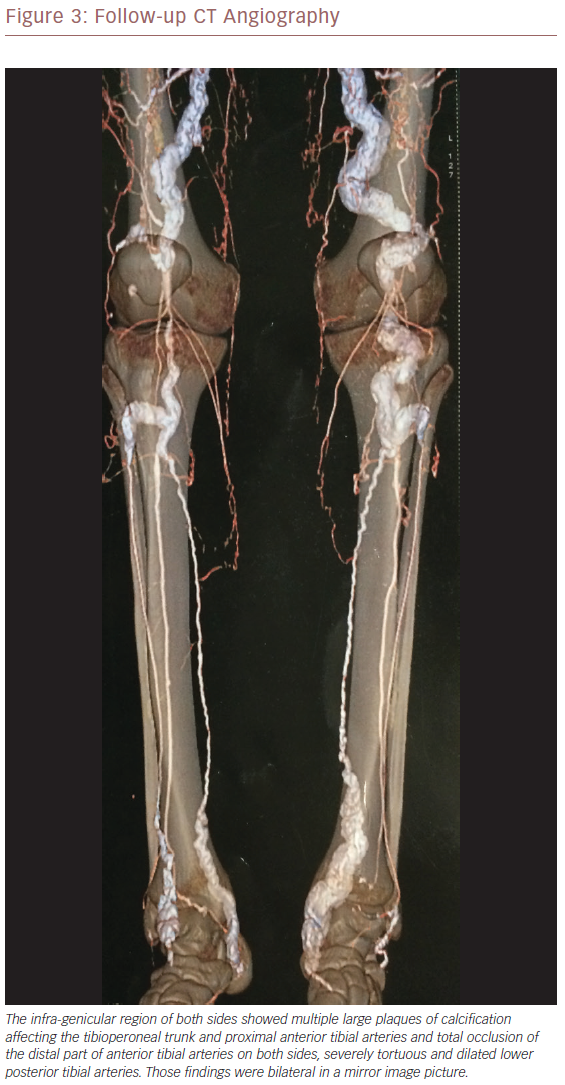
Follow-up CT angiography 12 months later showed a mirror-image picture with no major changes from the previously noted heavy calcification. There was more dilatation, kinking of the lumens and early affection of CFA and proximal SFA (Figures 2 and 3). As there had been no improvement since initial presentation, open surgical revascularisation or angioplasty were considered. Because his condition is deteriorating, revascularisation is mandatory.
Discussion
The main cause of lower limb chronic ischaemia is atherosclerosis, caused by the formation of atheromatous plaques in the arteries. Known risk factors for atherosclerosis include family history, being male, old age, smoking, diabetes, dyslipidaemia and hypertension.11 If claudication pain occurs in patients who have none of these risk factors, other, less common aetiologies should be considered.12
Non-atherosclerotic peripheral artery diseases are a diverse group of disorders with different pathophysiologies, clinical manifestations and treatments. They include popliteal artery entrapment syndrome, cystic adventitial disease, iliac artery endofibrosis, fibromuscular dysplasia, Buerger’s disease, chronic exertional compartment syndrome, and medium and large vessel vasculitis, including Takayasu’s arteritis, giant cell arteritis and Behcet’s disease. The diagnostic approach to patients presenting with intermittent claudication must consider atherosclerotic as well as non-atherosclerotic causes of peripheral artery disease (PAD).13
The debate concerning the pathological process of MC and its association with intimal calcification in atherosclerosis goes back to 1903, when Johann Georg Mönckeberg first described medial calcification in the arteries of older people. He described stages of disease progression which could be concluded to be stages of atherosclerotic plaque formation.14
The histopathology of PAD and the accompanying calcification are poorly defined, and it is not known whether this varies according to different risk factors. The majority of arteries in patients with PAD have a vascular lesion that is distinct from atherosclerosis, suggesting a different pathogenesis, and the main bulk of vascular calcification in the lower extremities is medial rather than intimal.15
Michelle et al. reviewed the literature on medial calcification and found reports of internal elastic lamina calcification.2 This was a good demonstration of the variability of atherosclerotic calcification in the involved layers of the arterial wall which has been misinterpreted as Mönckeberg’s sclerosis in the literature.
In articles that attempt to distinguish between atherosclerotic lesions and pure medial calcinosis on the basis of location and the pattern of calcifications, there was an overlap between intimal or medial which involved the internal elastic lamina border between those planes. When appropriately stained, lipid material was present, confirming atherosclerosis in some of the most densely calcified medial arteries. The histomorphology results were consistent for both atherosclerosis and MA. The calcific process included expression of osteoprotegerin, TNF-related apoptosis and mRNA expression patterns which were similar for both atherosclerosis and MA. Thus, MA seems to be a variant of advanced, calcified atherosclerosis with little inflammation and no clear evidence of an independent, non-atherogenic process.6,16
Zhang et al. studied the distribution of atherosclerotic plaques in the arterial beds of the common femoral artery bifurcation. Plaque tends to be located in areas opposite the carina in SFAs. Normal arteries were more common in profunda femoris arteries than in common femoral arteries and SFAs.17
In our patient’s angiography, the profunda femoris arteries were heavily loaded with calcification and the common femoral artery was spared, which does not support atherosclerotic pathology.
Radiographically, medial calcinosis presents as areas of linear calcifications in soft tissue.18 Goebel and Füessl identified medial calcification in the legs using X-ray without histological confirmation.19 However, attempts to distinguish and quantify medial calcification using radiological patterns or by examining peripheral arteries are problematic because of poor specificity, the existence of atherosclerosis in some peripheral arteries, the lack of validation and the semi-quantitative nature of the measurements.20
In our case, plain radiography was more consistent with criteria mentioned in the literature for the diagnosis of Mönckeberg’s arteriosclerosis (Figure 1). Our patient had no risk factors for atherosclerosis. He was thoroughly examined and investigated for all possible causes of hyperkalemia, dyslipidaemia or for other possible causes. The inherited condition PXE was a more appropriate aetiology to consider.
In 2014, Leftheriotis et al. studied the impact of calcification on arterial wall stiffness in PXE.21 PXE is an autosomal recessive disease caused by mutations in the ABCC6 gene and is characterised by progressive calcification of the elastic fibres in the skin, retinal Bruch’s membrane and the medial layers of large and medium-sized peripheral arteries. People with PXE develop cardiovascular events and complications, including PAD, at a younger age than the normal population, but they do not have obvious cardiovascular risk factors. Our patient could have a variant presentation of PXE.
The absence of an accepted vascular calcium grading scale hampers the clinical assessment of the safety of endovascular devices in the treatment of calcified vessels.22 Flore et al. quantified non-coronary vascular calcification in asymptomatic patients at low to intermediate cardiovascular risk using a colour Doppler ultrasound-based score – the carotid, aortic, lower limbs calcium score (CALCS).23 They found that the CALCS correlated with other validated markers of subclinical atherosclerosis, such as carotid intima-media thickness and arterial stiffness. They concluded that CALCS may be a reliable alternative to the traditional coronary artery calcification score in the evaluation of peripheral calcium load and it allows a better stratification of patients. Hendriks recommends different methods of assessing calcification other than Agatston scoring because arterial calcification is an actively regulated process with associated detrimental effects, and the risk factor profile associated with MA differs from that of intimal arterial calcification.24
The presence of MA does not necessarily mean revascularisation cannot be performed. In some cases, revascularisation is possible, and will involve anastomosis and avoidance of clamping the artery, but in other situations, revascularisation is impossible due to intense calcification.25
In 2015, Castling et al. reported their experience of using vessels affected by MA with medial calcification in an osseocutaneous radial forearm free flap reconstruction.26 They found that the anastomosis of affected arteries had no impact on flap viability. This suggests that there is the possibility of open revascularisation in these patients.
Conclusion
Although rare, Mönckeberg’s calcification should be considered as a potential cause of peripheral vasculopathy in a patient presenting with peripheral vascular disease with undefined aetiology in the absence of known risk factors. There is great diversity regarding arterial affection. Further studies are needed for clinicians to have a better understanding of the disease as a separate pathological entity. It would be useful to differentiate between the response of medial calcification to angioplasty, or even open revascularisation and that of predominant intimal calcification to create a treatment strategy for this condition.